[기술백서] AAV8 벡터 제조 플랫폼 구축
2026.05.13





미국 텍사스에 있는 CGT CDMO 전문기업 마티카 바이오테크놀로지(이하 마티카 바이오)는 글로벌 바이오 공정기술 솔루션 기업 사토리우스(Sartorius)와 AAV8 유전자치료제 제조 플랫폼을 구축했다. 마티카 바이오는 싱글유즈시스템(Single use system)과 디지털 분석을 결합해 AAV8 벡터 제조 과정을 표준화하고, 생산 규모를 50리터 규모까지 확장하는 전략을 기술백서에서 공개했다.
바이럴 벡터(Viral Vector)는 DNA나 RNA와 같은 유전물질을 세포에 주입하기 위해 개발된 운반체다. 유전자치료제 개발에 필수 요소다. 유전자치료제에 사용되는 바이럴 벡터 중 시장을 주도하는 것은 AAV(adeno-associated virus, 아데노 연관 바이러스)다. 전 세계 3000명 이상의 환자가 노바티스의 척수성근위축증 치료제 ‘졸겐스마(Zolgensma)’, 유전성 망막질환 치료제 ‘럭스터나(Luxturna)’ 등 AVV 기반 유전자치료제를 투여 받았고, 700개 이상의 치료제 개발 연구가 진행 중이다. 특히 아데노 연관 바이러스의 일종인 AAV8은 안정성과 유전자 전달 효율이 높아 근이영양증, 혈우병, 망막질환 임상시험에서 광범위하게 활용되고 있다.
유전차치료제 시장이 커지면서 바이럴 벡터 수요도 급격하게 늘어나고 있다. CGT(세포·유전자치료제) CDMO 기업은 환자에게 안전하게 사용할 수 있는 높은 품질의 바이럴 벡터를 균일하게 생산해야 하는 과제를 안게됐다. 여러 고객사의 요구를 맞추기 위해 매번 처음부터 제조 공정을 새로 설계하는 방식을 극복하려면 플랫폼 구축이 필요하다.
마티카 바이오는 사토리우스와 함께 50리터 규모까지 확장 가능한 AVV8 제조 플랫폼을 구축하고, 모든 공정에서 균일한 품질의 벡터 생산이 가능한지 검증했다.
세포·유전자치료제 초기 개발 단계에서는 1~10리터의 소규모 바이럴 벡터가 필요하다. 50리터 규모는 환자에게 투여할 수 있는 GMP 수준의 임상용 의약품을 제조할 수 있는 체계를 갖추었으며, 향후 상업화 생산으로 나아가기 위한 핵심 기술 기반이 마련되었음을 의미한다.
소규모 실험용부터 50리터까지 업스트림 공정
공정 개발의 첫 단계는 세포를 배양하고 바이럴 벡터를 생산하는 단계다. 이때 가장 고려해야 할 부분은 생산 규모다. 임상 단계나 규제기관 인허가를 위한 소량 생산을 넘어 대규모 환자 치료에 사용할 수 있도록 대량 생산이 가능해야 한다. 업스트림(Upstream) 공정은 이러한 요구를 충족하기 위해 생산 규모를 확장해 나가는 스케일업 과정이라고 할 수 있다.

<사토리우스 사의 Ambr®250>
마티카 바이오는 사토리우스의 최신 세포배양(바이오리액터) 시스템인 Ambr®250을 사용해 ▲수소이온농도지수(pH) ▲용존산소(DO) 및 이산화탄소(CO₂) 농도 ▲온도 등 다양한 조건을 테스트했다. 실험설계(Design of Experiment, DoE) 소프트웨어를 활용해 수십 가지 조건을 체계적으로 분석해 안정적인 설정값과 공정이 가장 안정적인 변수를 찾아냈다.
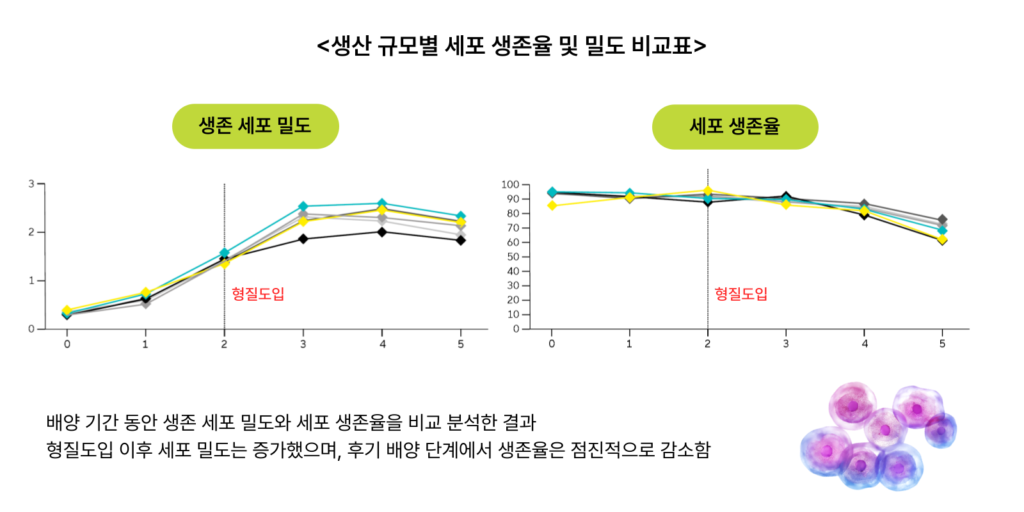
이후 공정 규모를 단계적으로 늘려나갔다. 250mL 소규모 실험 → 2L 생물반응기 → 50L 생물반응기 순서로 스케일업을 진행했고, 생산 규모가 커져도 세포의 생존율뿐만 아니라 물리적, 생물학적, 화학적, 미생물학적 특성이 일관되고 안정적으로 유지되는 것을 확인했다. 이는 소규모에서 얻은 공정 데이터를 대규모 생산에 그대로 적용할 수 있다는 것을 의미한다.
불순물 제거를 위한 다운스트림(정제) 공정
다운스트림(Downstream, 정제공정)은 배양을 통해 생산된 세포에서 불필요한 부산물을 제거하고 바이럴 벡터만 추출하는 과정이다.
바이럴 벡터를 생산 할 때 유전물질이 채워지지 않은 빈 캡시드(Empty Capsid)가 생성될 수 있다. 빈 캡시드가 포함되어 있는 경우 약물의 정확한 용량을 측정하기 어렵고, 환자에게 비특이적 면역반응이 일어날 수 있어, 생산 과정에서 완전히 제거하는 것이 중요하다.
정제공정은 큰 입자와 세포 잔여물 제거하는 여과(Filtration)와 크로마토그래피 레진(resin)을 사용해 미세 분순물을 제거하는 크로마토그래피(chromatography) 단계를 거친다.
마티카 바이오는 여러 종류의 접선유동여과(Tangential Flow Filtration, TFF) 필터(membrane)을 비교 평가해 최적의 소재를 선정했다. 크로마토그래피 단계에서는 사토리우스의 CIMmultus PrimaS를 사용해 충전 캡시드 비율을 약 3.2배 높이는 데 성공했다. 정제 후 충전 캡시드 비율은 약 70%에 달했다.
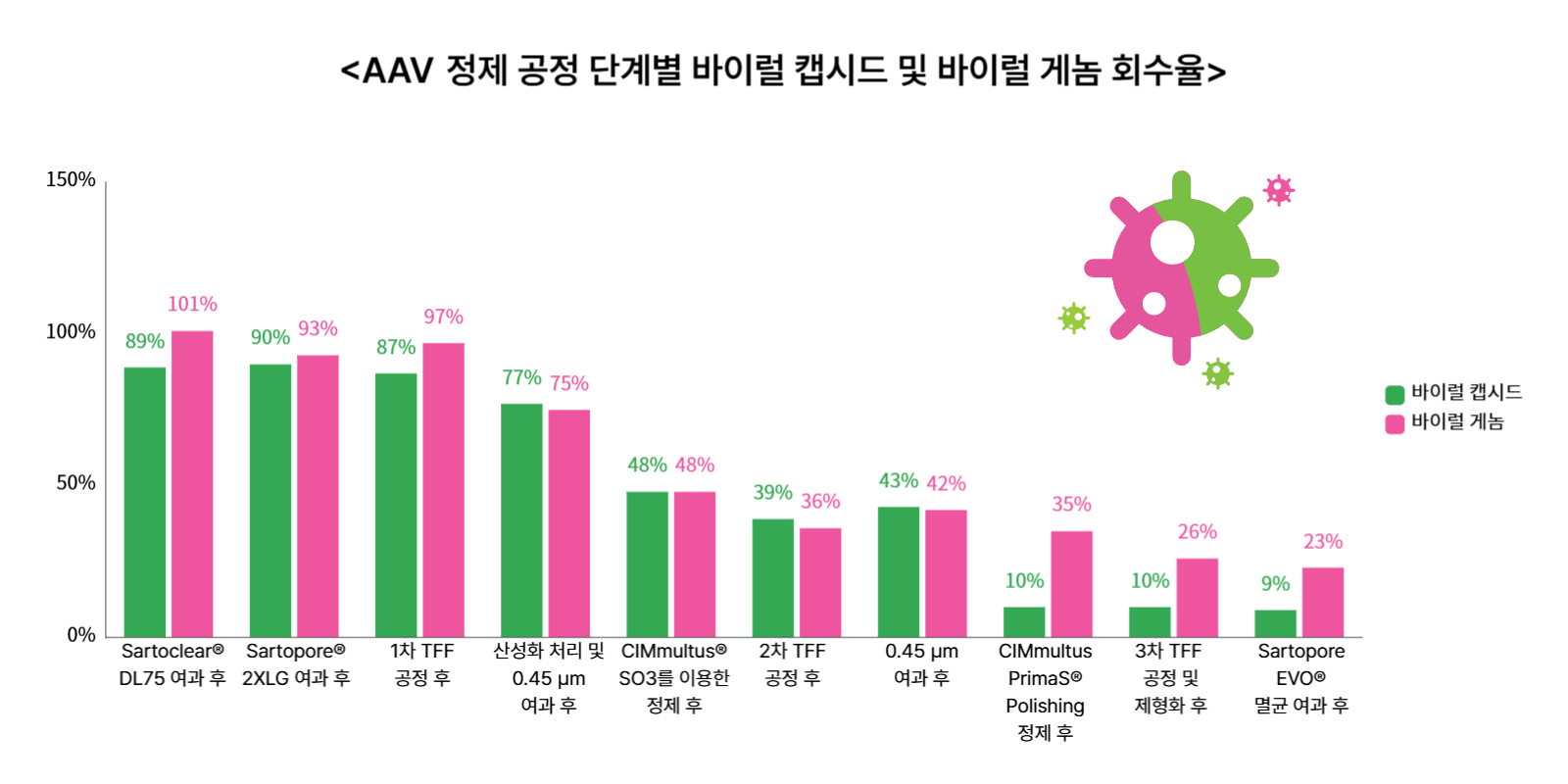
전체 공정의 최종 회수율은 바이럴 게놈 기준 23%, 캡시드 기준 9%로, 이는 현재 글로벌 업계 수준이다. 캡시드 회수율이 상대적으로 낮아 보이지만, 이는 빈 캡시드가 효과적으로 제거되었기 때문이다. 즉, 전체 캡시드 수는 줄었지만, 실제로 유전자를 포함한 유효한 바이러스의 비율이 높아져, 제품의 순도(Purity) 측면에서는 오히려 긍정적인 결과로 볼 수 있다.
※ AAV 제조 과정에서 회수율 측정 지표
1. 바이러스 게놈(VG) 회수율
ddPCR(드롭렛 디지털 중합효소 연쇄반응)을 활용해 정량화된 DNA 함유 입자의 회수율을 측정한다. 마티카 바이오의 공정에서 23%의 VG 회수율은 다운스트림 공정 전반에 걸쳐 DNA 함유 물질이 효율적으로 유지되고 있음을 반영한다. DNA를 포함하는 AAV 입자(기능성 캡시드 및 부분적으로 채워진 캡시드 모두)를 나타낸다. 종종 업스트림 공정에서 생산된 산출물의 측정 지표로 사용되며, 다운스트림 정제 과정에서 빈 캡시드가 제거되는 영향이 상대적으로 적다.
2. 총 캡시드 회수율(Total Capsid Recovery)
전체 AAV 캡시드 입자(유전자를 포함하는 완전 캡시드 및 유전자가 없는 빈 캡시드)를 측정한다. 9%의 캡시드 회수율은 정제 후 남아 있는 총 물리적 입자의 비율을 나타낸다.
마티카 바이오는 정제 단계에서 전체 캡시드의 농도를 약 3배로 높이고, 전체 캡시드 함량을 약 70% 수준으로 달성했다. AAV 생산 과정에서는 자연스럽게 상당량의 빈 캡시드가 생성되고, 크로마토그래피 단계에서 빈 캡시드는 제거된다. 이 단계를 거치면서 제품의 효능, 투여량 정확도 및 규제 당국의 승인 가능성이 향상된다.
총 캡시드 회수율은 제품의 손실이 아닌 기능이 없는 빈 캡시드를 선별적으로, 성공적으로 제거한 결과를 의미한다. 그래서 AAV제조 과정에서 회수율은 VG 회수율과 총 캡시드 회수율을 함께 살펴봐야 한다.
제조시간과 비용을 줄이는 싱글유즈시스템
이번에 구축한 픗랫폼의 또 다른 특징은 싱글유즈시스템(Single use system)을 적용한 것이다. 업스트림부터 다운스트림까지 전 공정에 단회용 장비를 적극 활용했다.

싱글유즈시스템은 고정된 용기와 배양기 등을 사용하는 멀티유즈(Multi-Use)시스템과 달리 사용 후 폐기하기 때문에 배치(batch) 간 세정 작업이 필요 없고, 교차 오염 위험도 크게 줄어든다. 바이럴 벡터 생산에서는 생물안전 요건이 엄격하기 때문에, 이 같은 특성은 매우 중요한 장점이다. 업계 조사에 따르면 단회용 시스템을 도입하면 제조 기간을 최대 40%, 설비 투자 비용을 최대 60%까지 줄일 수 있다.
규제기관 승인에 적합한 GMP 적합성
규제기관은 단순히 결과 데이터만 보는 것이 아니라, 공정 전체의 일관성과 통제력을 평가한다. 마티카 바이오는 개발 초기 단계부터 GMP(우수의약품 제조·관리 기준) 환경에서의 적합성을 고려해 각 공정 단계를 설계했다.
사토리우스 장비를 선택한 이유 중 하나도 검증 패키지, 표준작업절차서(SOP), 전자 배치 기록 호환성 등 규제 대응 인프라를 갖췄기 때문이다. 이를 통해 개발 단계에서 GMP 생산으로의 전환 시 추가적인 재검증 부담을 줄일 수 있었다.
이번에 구축한 AAV8 제조 플랫폼은 AAV2, AAV5, AAV9 등 다른 아데노 연관 바이러스에도 동일하게 적용할 수 있다. 마티카 바이오는 사토리우스와의 이번 협업으로 특정 프로젝트 하나를 완수한 것이 아니라, 앞으로 다양한 고객사의 유전자치료제 프로젝트를 더 빠르고 안정적으로 지원할 수 있는 전략적 자산을 확보했다. CGT CDMO에게 플랫폼은 단순한 기술이 아닌, 경쟁력의 핵심이다.

< 마티카 바이오 연구원들이 바이럴 벡터 농도 분석 결과에 대해 논의하고 있다>
플랫폼 방식을 도입할 경우 고객사는 크게 세가지 부분에서 효과를 볼 수 있다.
첫째, 치료제 개발 시간과 비용을 줄일 수 있다. 이미 검증된 공정 조건과 분석법을 사용할 수 있어 빠르게 프로젝트를 진행할 수 있다.
둘째, 기술이전(Tech Transfer)이 수월해진다. 표준화된 장비와 절차를 사용하기 때문에, 다른 시설이나 파트너사에 공정을 이전할 때 발생하는 오류와 재교육 부담이 줄어든다.
셋째, 규제 대응에 유리하다. 각 공정 단계의 성능 데이터가 이미 축적되어 있어 미국 식품의약국(FDA), 유럽의약품청(EMA) 등 규제기관에 제출할 자료를 준비하는 시간도 단축된다. #